 软件星级:3分
软件星级:3分

2、开始安装;

3、选择我同意相关协议;
4、选择安装路径;
5、正在安装;
6、安装完成;
7、如图所示,打开文件安装位置;
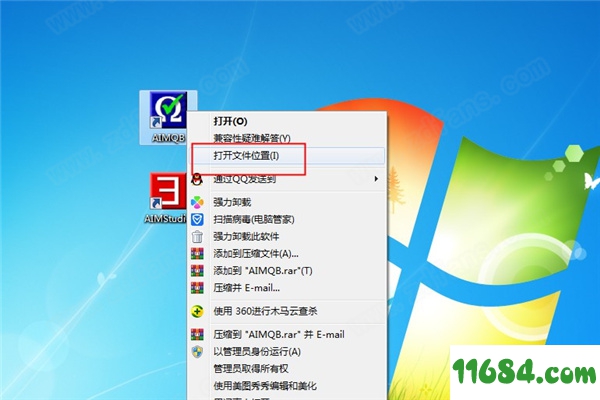
8、找到破解补丁;
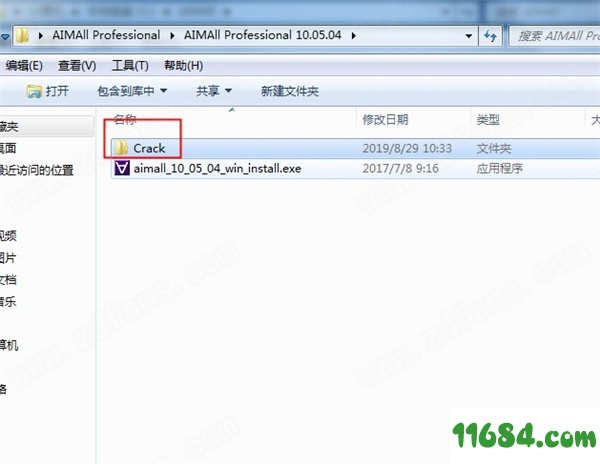
9、把补丁复制进去即可;
10、打开软件。
2、支持各种原子轨道;
3、加快计算和做事情;
4、支持不同的操作系统。
在Windows,MacOSX和Linux上完全正常运行。
易于使用的GUI和命令行界面,用于设置和运行部分或完整的AIM分析。
完整的GUI支持,可轻松设置和运行多个串联波函数的AIMAll计算(“批处理模式”)。
全自动,可靠,稳健,准确,相对快速,高效地计算电子密度拓扑和所有原子特性。
如有必要,自动调整原子积分方法和/或原子积分参数,以始终实现良好的原子积分精度。
支持传统的AIM波函数文件(.wfn),扩展的AIM波函数文件(.wfx)和高斯(g03或g09)格式的检查点文件作为输入。
无限的波函数大小,即无限数量的原子,MO和基函数。
正确处理各种波函数类型:限制HF/KSDFT,限制开壳HF/KSDFT,无限制HF/KSDFT,限制MP2/CCSD/等,无限制MP2/CCSD/等,CASSCF,CIS,磁性(GIAO/CSGT)扰动HF/KSDFT等
将高斯格式化检查点文件自动转换为扩展或传统AIM波形函数文件。
自动验证有时有问题的传统波函数文件和扩展波函数文件。
对于任何类型的计算,支持波函数扩展为S,P,D,F,G和H高斯基函数类型。
当波函数包括用于ECP建模的核心电子的附加核心密度函数数据时,支持使用有效核心势(ECP)的计算得到的波函数。
信息性,可定制的日志窗口显示计算进度。
共享内存,多处理器/多核支持所有重要的计算步骤。
能够使用多个共享内存处理器/内核一次无缝地计算多个原子。
电子密度分布的自动和彻底的拓扑分析,包括具有非常复杂拓扑的那些。
丰富的电子密度临界点和路径描述符,包括所有CP(和核)的电子密度,BCP的椭圆度,所有CP的能量密度,所有CP(和核)的总静电势,所有CP的应力张量数据,键合路径长度,键合路径角度等。
自动计算和保存完整的分子图形可视化。
所有电子密度临界点和路径属性都有用地汇总在易于阅读,自描述的文本文件和相应的可视化文件中。
全自动分析拉普拉斯算子的电子密度分布的拓扑特征,包括所有临界点及其性质以及原子图路径。
各种功能的交互式拓扑分析,包括维里场,磁感应电流密度分布和单个分子轨道。
自动,完整地设置和提交所有原子集成计算。
同时原子盆地和原子表面性质计算。
提供多种独立的原子集成算法,用于内部验证和处理特别困难的情况。
原子电荷,偶极矩贡献,四极矩,径向矩和径向畸变矩。
原子动能,势能,总能量和拉格朗日。
完整而准确地实现IQA(交互量子原子),包括Vee(A,B)术语,用于Hartree-Fock,LSDA和B3LYP以及M062X波函数。
原子体积和原子表面积,包括原子间表面区域(等距密度表面)和原子等密度表面区域。
原子电子定位指数(LIs)和离域指数(DIs,债券订单)。
计算原子静电相互作用能的所有成分。
可选计算原子Ehrenfest力和原子Feynman力。
可选计算对库仑,交换和总双电子能量的原子贡献。
可选计算原子磁响应特性,如净电流,磁化率,NMR屏蔽张量(包括NIC)等。
原子笛卡尔矩的电子密度分布通过5阶。
原子球谐波矩的电子密度分布为任意阶数(默认情况下高达l=5,根据要求可提高)。
计算原子间表面性质和原子间表面文件的写入,以便在以后的计算中可视化和可能的重复使用。
计算原子等密度表面(IDS)特性,包括面积和静电势特性。
用于研究开壳系统的原子电子自旋种群和其他自旋特性。
原子源对所有电子密度临界点和所有原子核的电子密度的贡献。
所有原子属性都在一个易于阅读,自描述的文本文件和相应的可视化文件中进行了有用的汇总和制表。
二、可视化功能
在Windows,MacOSX和Linux上完全正常运行。
现代3D(OpenGL)可视化应用程序(AIMStudio),用于交互式,完全可自定义的3D窗口显示和所有AIMAll计算结果的表格显示。
显示可定制的分子图,单独或与任何相关的原子属性,电子密度临界点属性,等高线图,浮雕图,等值面(原子和分子),原子间表面,电子密度的拉普拉斯算子的临界点和性质等一起显示。
在多个3D窗口和表格中显示多个分子的结果,以便在任务之间轻松切换并轻松比较结果。
可分类,可复制,可打印的表格,适用于所有原子属性,键临界点属性,环临界点属性,笼关键点属性,非核吸引子属性和电子密度临界点属性的拉普拉斯算子。
以各种流行格式保存3D窗口的高分辨率图像文件,包括PNG,TIFF,JPEG,BMP和PDF。
打印3D窗口的高分辨率图像。打印预览可用。
易于计算和可定制的轮廓图3D显示(可按值分类,可根据用户指定的位置和值标签的外观),浮雕图(可按值分类),2d彩色图和等值面,用于各种功能,包括电子密度,拉普拉斯算子的电子密度,各种能量密度,总静电势(ESP),电子自旋密度,单个分子轨道,磁感应电流密度大小,Johnson等人的非共价相互作用(NCI)图等。
能够显示磁感应电流密度分布的矢量箭头图。
能够将一个函数颜色映射到另一个函数的等值面上,例如将ESP映射到电子密度等值面上,具有用户可指定的布局和映射函数的值标签的外观。
丰富的3D窗口显示选项,包括可调节的背景颜色,可调节的球体半径和每种对象类型的路径/线宽,每种对象类型的可调节字体,每种对象类型(包括文本)的可调颜色和透明度,透视查看,抗锯齿,可调节照明等
在3D窗口中单独或按类型隐藏对象。
任何一组选定的核,BCP,RCP,CCP,NNACP或Ghosts的交互式距离表。
在视觉上区分弱粘合路径。
完全可定制的文本文件查看器,用于查看和搜索详细的结果文件
用户定义的3D窗口文本注释。
可定制显示3D窗口中的一个或多个原子间表面和/或原子等密度表面和/或原子盆路径组。
从同一波函数派生的任何可视化文件都可以在同一个3D窗口中轻松显示。
可定制的交互式3D显示和表格显示关键点,关键点属性和拉普拉斯电子密度的原子图路径。
对AIMAll标准操作模式或AIMAll Professional操作模式的计算功能没有限制。对于AIMAll标准操作模式,可视化相关功能和共享存储器多处理器支持限制为12个原子或更少,400个原语或更少。使用AIMAll Professional操作模式对可视化相关功能或共享内存多处理器支持没有限制。
2、可以使用通过典型的Kohn-Sham DFT(KS-DFT)方法计算得到的波函数吗?
是的,使用KS-DFT波函数计算和解释大多数QTAIM属性类似于Hartree-Fock波函数,使用合适的KS-DFT方法可能更准确。为了计算基于原子DFT的交换相关能量和附加的IQA原子能量,目前唯一支持的KS-DFT方法是LSDA和B3LYP和M062X。必须使用
3、AIMAll是否支持使用多个处理器/核心?
是的,支持使用多个处理器(仅限共享内存)可选地运行AIMExt和AIMInt作业)是可用的(非小波浪功能需要AIMAll Professional)。还支持使用多个处理器同时运行多个AIMInt作业(即,一次计算多个原子)(非小波函数需要AIMAll Professional)。多处理器选项可通过AIMQB对话框中的“处理器”和“一次原子”字段或通过AIMQB的“-nproc”和“-naat”命令行参数指定。注意,对于小波函数最有效地使用多个处理器是一次计算多个原子(例如,-nproc = 3 -naat = 3)。对于大波函数,每个原子计算使用多个处理器变得有效(例如,-nproc = 3 -naat = 1)
4、如何用AIMAll计算原子磁响应特性?
计算磁化张量和NMR屏蔽张量的原子贡献要求在AIMQB的“磁响应特性”组合框中选择CSGTB,IGAIM或GIAO,以及使用CSGT磁波函数文件或GIAO磁波函数文件。
传统AIM格式(.wfn)的CSGT磁波函数文件可以由Gaussian使用以下关键词产生:nmr = csgt iop(10/21 = 1,99 / 6 = 200)output = wfn
扩展格式的CSGT 磁波函数文件(.wfx)可以由Gaussian 09 Revision B.01及更高版本使用关键字:nmr = csgt output =(wfx,csgtcx)生成
必须手动将关键字“CSGT”(不带引号)添加到传统AIM格式的CSGT磁波函数文件的第2行(NUCLEI之后)的末尾,以便AIMAll知道它是CSGT波函数文件。
CSGT磁波函数可用于CSGTB和计算AIMAll中原子磁响应特性的IGAIM方法。AIMAll中的CSGTB方法与Gaussian中实现的CSGT方法相同。AIMAll中的IGAIM方法(使用QTAIM原子)与高斯中的IGAIM方法不同,并且对于较小的基组,产生略微不同的总结果。
传统AIM格式(.wfn)的GIAO磁波函数文件可以由Gaussian使用以下关键词产生:nmr = giao iop(10/21 = 1,99 / 6 = 300)
扩展格式(.wfx)的GIAO磁波函数文件可以由Gaussian 09 Revision B.01及更高版本使用关键字产生:nmr = giao output =(wfx,giaocx)
关键字“GIAO”(不带引号)必须是手动添加到传统AIM格式的GIAO波函数文件的第2行(NUCLEI之后)的末尾,以便AIMAll知道它是GIAO波函数文件。
AIMQB也可以通过打开由nmr = giao计算得到的高斯格式检查点文件自动生成扩展AIM格式(.wfx)或传统AIM格式(.wfn)的GIAO磁波函数文件。
5、sum文件中有关“重要”或“潜在重要”集成错误的消息是什么意思?
理想情况下,计算的原子L(A)值应为零,并且分子中所有原子的任何计算的原子性质的总和当然应该等于已知的分子值。在实践中,总会存在一些数值积分误差,通常与原子表面的复杂性有关。
如果发生以下任何一种情况,“警告!重要...集成错误...”消息将写入总和文件:
原子A的计算L(A)值的大小> 0.01 au。
所有计算的L(A)值之和的大小> 0.01 au。
实际分子电荷与所有计算的原子电荷之和q(A)之差的大小> 0.01 au。
如果出现以下任何一种情况,“Note。潜在重要...集成错误...”消息将写入sum文件:
原子A计算的L(A)值的大小> 0.002 au和< 0.01 au。
所有计算的L(A)值之和的大小是> 0.002 au和< 0.01 au。
实际分子电荷与所有计算的原子电荷之和q(A)之差的大小是> 0.002 au和< 0.01 au。
“有效” 的0.01 au和“潜在有效”积分误差的0.002 au 的阈值是合理的,但根据一个人的目的,可能过于保守或不够保守。
注意,由于误差的消除,计算的原子L(A)值可以(但不可能)在量值上非常小。
建议用户自己监测计算的原子特性的总和以及各个计算的L(A)值,并准备好在必要时使用更高的正交和/或不同的原子积分算法重新计算“问题原子”。
6、何使用AIMAll计算分子中偶极子极化能力的张量?
分子偶极极化率张量的分量可以被计算为分子偶极矩分量相对于所施加电场的分量的一阶导数,该导数在零场点处被评估。分子中的原子也是如此。为了计算原子偶极子衍生物,必须使用数值差异,因为原子表面(由电子密度分布限定)取决于电场。电子表格可能会有所帮助。
基本步骤是:
对感兴趣的分子进行常规的零场ab initio计算并生成相应的波函数文件,例如molecular_0.wfn。
在molecular_0.wfn上运行AIMAll以产生molecule_0.sum,其将包含零场点处分子的所有原子偶极矩。
在相同的几何形状,理论水平和基础上进行另一个ab initio计算分子,但是在存在沿+ X轴施加的小(例如0.0025 au)电场并产生相应的波函数文件时,例如molecular_0025_x.wfn 。
在molecular_0025_x.wfn上运行AIMAll以产生分子_0025_x.sum,其将包含在施加的电场存在下分子的所有原子偶极矩。
对于分子中的每个原子,计算原子极化率张量的XX,YX和ZX分量,作为原子偶极矩的X,Y和Z分量的变化除以沿X轴的外加电场的变化,即0.0025 au。
重复步骤3-5,但是沿着+ Y轴施加电场,以计算原子极化率张量的XY,YY和ZY元素。
重复步骤3-5,但是沿着+ Z轴施加电场,以计算原子极化率张量的XZ,YZ和ZZ元素。
考虑沿-X,-Y和-Z轴施加电场重复上述步骤。
考虑用较小(例如0.001 au)和较大(例如0.005 au)场强重复上述步骤。
7、如何在AIMStudio的3D窗口中使对象半透明?
通过使用对应类型的相应颜色对话框中的“Alpha通道”字段,可以在AIMStudio的3D窗口中使任何类型的对象(包括文本)半透明。Alpha通道值的范围可以从255(不透明)到0(不可见)。大多数对象类型默认是不透明的。
8、如何在AIMStudio中获得更平滑的线条/路径/曲线/文字?
抗锯齿可能(取决于计算机图形卡的质量)改善路径和点(以及可能的文本)的外观(平滑度)。默认情况下启用抗锯齿功能,但可以通过“查看 - >抗锯齿”菜单项关闭。对于某些低端图形系统,抗锯齿可能效果不佳。在许多情况下,可以使用图形系统的“控制面板”GUI调整图形系统的抗锯齿支持。或者,您可以选中“视图 - >管状线,曲线和路径”菜单项,它将使用圆柱段而不是光栅化线段绘制线条,路径和曲线。使用圆柱段的绘制线,路径和曲线在屏幕和图片(包括高分辨率)中看起来都很好但可能很昂贵。
9、如何在AIMStudio中的3D窗口中放大和缩小对象?
以下任何一种都可用于缩放:
使用鼠标滚轮或*板或魔术鼠标滚动机制。
使用键盘上的F5和F6键
按住鼠标右键并按住Shift键水平移动鼠标
选择“View-> Molecule Positioning-> Tools ...”菜单项以使用“Molecule Positioning Tools”对话框。
10、如何在AIMStudio中的3D窗口中旋转对象?
可以使用以下任何一种旋转:
通过按住鼠标左键在窗口中左右移动鼠标光标,围绕屏幕Y轴旋转。
使用向左和向右箭头键盘键围绕屏幕Y轴旋转。
通过按住鼠标左键在窗口中来回移动鼠标光标,围绕屏幕X轴旋转。
使用向上和向下箭头键盘键围绕屏幕X轴旋转。
通过按住鼠标右键在窗口中左右移动鼠标光标,围绕屏幕Z轴旋转。
使用Page Up和Page Down键盘键围绕屏幕Z轴旋转。
选择“View-> Molecule Positioning-> Tools ...”菜单项以使用“Molecule Positioning Tools”对话框。
11、如何在AIMStudio中的3D窗口中左/右/上/下移动(平移)对象?
以下任何一种都可用于翻译:
通过按住鼠标中键(或可能是滚轮)在窗口中左右移动鼠标光标,沿着屏幕X轴移动。
通过按住鼠标左键并按住Shift键,在窗口中左右移动鼠标光标,沿屏幕X轴移动。
按住Shift键,使用左右箭头键盘键沿屏幕X轴移动。
通过按住鼠标中键(或滚轮)在窗口中上下移动鼠标光标,沿着屏幕Y轴移动。
按住鼠标左键并按住Shift键,在窗口中上下移动鼠标光标,沿屏幕Y轴移动。
按住Shift键,使用向上和向下箭头键盘键沿屏幕Y轴移动。
选择“View-> Molecule Positioning-> Tools ...”菜单项以使用“Molecule Positioning Tools”对话框。
12、如何在我选择的空间中获得电子密度,电子密度的拉普拉斯算子和其他函数的值,而不仅仅是关键点?
运行程序AIMExt并使用主菜单选项(5),“输入坐标处的属性”。感兴趣的数据将在.extout文件中。
13、如何定位和表征维里场或其他功能的关键点?
在运行AIMQB时,始终自动计算电子密度的所有临界点。当运行AIMQB时,对于一些或所有原子,可任选地自动计算电子密度的拉普拉斯算子的临界点。对于其他电子密度和电子密度的拉普拉斯算子(例如,维里场)的函数,直接运行程序AIMExt并回答“你想要分析的主要功能是什么?”的问题。因此。然后使用主菜单选项(如(6))搜索主要功能的关键点。感兴趣的数据将在.extout文件中。
14、我可以在AIMStudio 3D窗口中隐藏单个原子,键合路径,BCP等吗?
是。右键单击任何核,BCP,RCP,CCP,NNACP,Ghost或其他对象范围将推送具有该对象特定的各种选项(位于菜单顶部)的上下文菜单。其中一个选项是“Cloak Object”。隐藏对象将隐藏它(使用更通用的术语“披风”而不是“隐藏”以允许将来可能的“隐形”的不同表示)。隐藏BCP也会掩盖其债券路径及其IAS EV路径。隐藏RCP也会隐藏其RCP到BCP路径。请注意,隐藏的对象无法通过右键单击它(因为它不再存在),但是所有隐藏的对象都可以通过“View-> Decloak All”菜单项去除。隐形修补对于非平面分子中的等高线图和其他2D图特别有用,
15、有没有办法让AIMAll对它产生的所有数值数据进行排序和/或过滤?
是。AIMStudio现在提供可定制的原子表, BCP表,RCP表,CCP表等,它们与相应的3D窗口交互,并允许用户以各种方式查看,排序,复制和打印属性数据。
16、.sum文件,.mgp文件,.extout文件和其他AIMAll结果文件中给出的长度,能量,电子密度等单位是多少?
AIMAll中的长度单位是原子单位(bohr)。AIMAll中的能量单位是原子单位(Hartree)。AIMAll中的电子密度单位是原子单位(e / bohr ^ 3)。如在每个.sum文件的顶部附近所述,除非另有说明,否则来自(和输入数据)AIMAll的所有输出数据都是原子单位。
17、AIMExt是否计算并打印Cremer和Kraka能量密度H(r)?
Cremer和Kraka能量密度H(r)= G(r)+ V(r)等于-K(r),即减去电子动能密度的哈密顿形式。K(r)由AIMExt在电子密度的关键点(例如BCP)计算,该数据可在.sum文件,.sumviz文件,.mgp文件和.mgpviz文件中获得。与所有其他CP属性一样,K(r)值可以在AIMStudio中以表格形式和/或在3D窗口中查看。与电子密度,电子密度的拉普拉斯算子等一样,函数K(r)本身可以使用AIMExt进行拓扑分析。可以使用AIMStudio显示K(r)的轮廓图,浮雕图和等值面。
18、AIMAll会计算原子电子自旋数吗?
是。原子数N_alpha(A),N_beta(A),N_total(A)和N_spin = N_alpha(A) - N_beta(A)被打印在原子积分结果文件(.int文件)的末尾附近以及其他原子电子旋转属性,如电子自旋动能,电子自旋矩等。原子自旋群也列在.sum文件和.sumviz文件的末尾,可在AIMStudio 3D窗口和Atom表中查看。这里显示了苯氧基中的原子电子自旋值N_spin(A)的图片。
19、AIMAll是否计算对NICS的原子贡献(“Nucleus Independent Chemical Shielding”)?
是的,只要你有适当的波函数来计算原子磁响应特性。要计算NICS屏蔽张量及其原子贡献,只需将“Ghost nuclei”的坐标添加到波函数文件中,以便计算要计算NICS贡献的每个点。对于每个鬼核,确保添加一个合适的核名称(必须以Bq开头,例如,Bq13),原子序数(0)和电荷(0.0),并确保增加核数量的值。鬼核补充说。
20、如何获得BCP与RCP,核心和BCP等之间的距离?我可以获得我感兴趣的关键点的角度和二面角数据吗?
对于通过键路径连接的原子核(NACP),原子核(NACP)与原子核(NACP)之间的距离与其共享键临界点的距离写入.mgp,.mgpviz,.sum和.sumviz文件并可以在AIMStudio中显示为BCP长度属性。键合路径角度和相应的几何键角也写入这些文件。涉及任何主要点对象(即,核,BCP,RCP,CCP,NNACP,Rho CP和Ghost的拉普拉斯算子)的距离,角度和二面角可以通过选择(点击)3D窗口中的对象球体从AIMStudio获得。通过“工具 - >选择表...”菜单项显示所选对象的距离表,角度表和二面角表。
